In an issue of Genome Biology in January 2018, Van Baak and colleagues analyzed the epigenome of monozygotic versus dizygotic twins. As you can see from the figure which was copied from the OpenAccess journal, dizygotic twins expose divergent methylation patterns in several genes (in red). Most remarkably is this for the DUSP22 gene which codes for a “dual specificity phosphatase”. On the other hand, monozygotic twins (in blue) showed almost no divergence, methylation rate ratio were very close to one, which would be on the diagonal line. 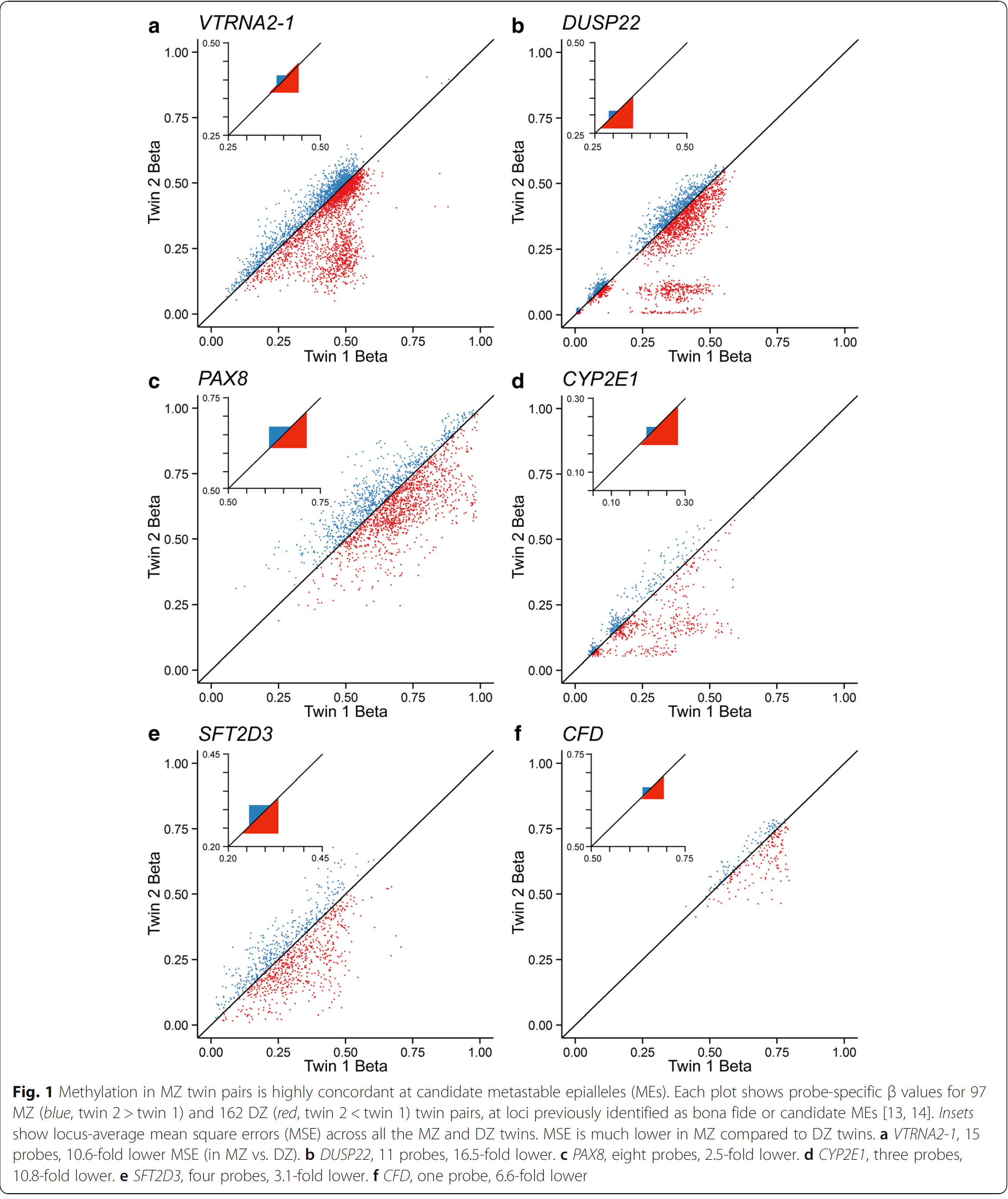
This lead van Baak et el. to look for the origin of this invariable methylation between two individuals. They found this supersimilarity between pairs of monozygotic twins is established at or before the stage when the twins become twins. This was fairly unexpected.
During their search the authors noted that subtelomeric regions are not only important targets for methylations, but at the origin of diseases e.g. tumour later in life.
Since the paper is very technical, it is difficult to read. The fact that monozygotic twins are not only identical in the genome, but extremely similar in their epigenome, too, is worth some labour with the text.